Protein Quantification
- dia-PASEF
- AQUA
- N15 & O18
- SILAC
- iTRAQ & TMT
- Label Free Quantitation
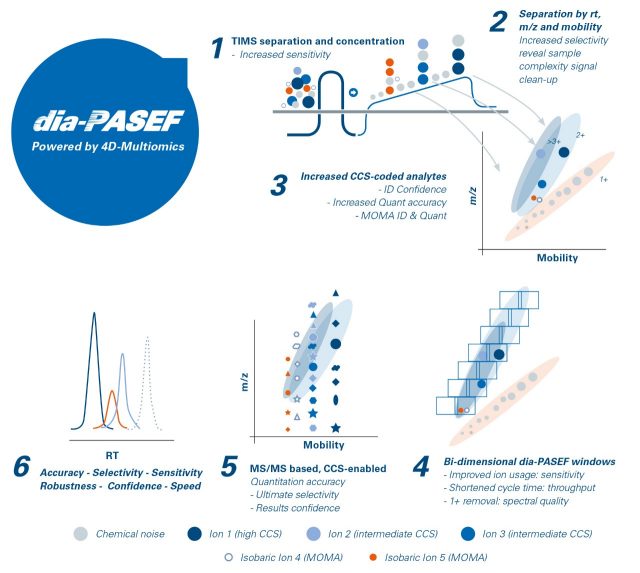
Data-independent acquisition (dia)-PASEF® is both more sensitive and selective than traditional DIA-MS approaches as it applies the PASEF® principle to combine the advantages of DIA with the inherent ion efficiency of PASEF®. Over the entire liquid chromatography-mass spectrometry (LC-MS)/MS dia-PASEF® run, a perfect data cuboid is created containing m/z, ion mobility (CCS), retention time and intensity. TIMS separation increases selectivity, excludes singly charged precursors from fragmentation and cleans up the sample by concentrating signals from noise. Making use of the correlation of molecular weight and CCS coded information from the dual-TIMS funnel, dia-PASEF® enables highly confident identification.
The facility is supported by the NIH shared instrumentation grant numbers
S10 OD016234 (Synapt-HDX-MS) and S10 OD021724 (LUMOS Orbi-Trap).
Please reference these grant numbers for publications resulting from data obtained from these instrumentations.
Funds from Moores Cancer Center of UC San Diego