Protein-Based Analysis
Protein Sequencing
RP-C18 (low pH): Low complexity (1-500 proteins) in gel or in solution
Protein Sequencing: Reverse Phase C18
Mass Spectrometry and Protein Sequencing
Most mass spectrometers that are used for protein sequencing are equipped with collision cells that are ideal for sequencing peptides that are under 4 KDa range. Mainly for this reason proteins need to be cut using proteases that will result in peptide fragments that on average are less than 4KDa. Once these peptides are generated they need to be fractionated before ionization into mass spectrometer. This is mainly to allow mass spectrometers time to analyze different peptides that are present in a mixture. The fractionation schemes can be carried out online or offline. The advantage of online schemes are that they require very little sample material which ultimately increases sensitivity of the system. For MALDI-TOF peptide sequencing, peptides will need to be fractionated offline for complex protein mixtures.
Reverse Phase (RP) C18 Resin Liquid Chromatography
The simplest fractionation strategy for low protein complexity samples is the reverse phase (RP) C18 resin liquid chromatography. In this separation scheme peptides bind the C18 resin based on their hydrophobicity. They are then eluted off the C18 resin using an organic reagent.
MUDPIT: Medium complexity In solution (1-1000 proteins)
Protein Sequencing: MUDPIT
Mass Spectrometry and Complex Protein Mixture Sequencing (1-3000 proteins)
MUDPIT (multidimensional protein identification technology) is the method of choice for complex protein sample analysis in which more elaborate separation techniques are needed. For MUDPIT analysis strong cation exchange resin (SCX), which binds positively charged compounds, is packed in tandem to the RP-C18 resin. SCX resin first binds all peptides before they encounter the C18 material. The peptides are then eluted off the SCX using ammonium salts in a stepwise manner, starting at low salt concentration step and ending with a high salt concentration step. After each salt step some peptides will be released from the SCX resin and will bind the RP-C18 material. This salt step is then followed by a cycle of organic gradient to elute off the peptides from the C18 resin and into the mass spectrometer for sequencing. For more complex samples more salt steps can be added to the method to increase the separation capability of the setup.
Recent method developments in peptide separation are using alternative separation strategies to SCX to improve peak separation and hence increase peptide identifications for Mudpit. One encouraging method is the use of "high-pH-reverse-phase" separation. Similar to low pH reverse phase separation techniques currently used in LC-MS, the high pH equally generates high peak resolution for peptides. We have observed that the use of this method in replacement of SCX in Mudpit analysis increases peptide identifications in similar Mudpit runs by the factor of two.

HpHRP: Off line High pH Reverse phase proteome fractionation coupled with RP-C18 low pH for high Complexity peptide separation.
Protein Sequencing: High pH Reverse Phase separation technique (HpHRP)
Multidimensional-liquid chromatography techniques are used to reduce the proteomic sample complexity prior to tandem mass spectrometry analysis to increase peptide identifications. The separation properties depend on the peak resolution in chromatographic dimensions. High pH reverse phase (HpHRP) has been shown to have a higher peak resolution when compared to strong cation exchange (SCX) separation and hence can replace SCX in multidimensional-liquid chromatography techniques (Gilar et al 2005).
Tryptic peptides extracted from proteomes of the interested organisms are fractionated on AKTA using the HpHRP strategy. The fractions are analyzed using LC-MS2 for peptide sequencing using standard low pH reverse phase.
Advantages of off-line HpHRP:
1) High loading capacity (milligrams of sample) for the detection of low abundant peptides.
2) High chromatographic peak resolution which reduces repeated sampling of same peptides in multiple fractions.

Data Analysis Tools (Software)
Protein Sequencing: Data Analysis
This facility offers a number of tools for automated and manual searches of tandem mass spectrometry results.
ProteinPilot
Is a commercially available software for automated database searches of tandem mass spectrometry data.
InsPecT
An online search engine for analysis of tandem mass spectrometry data. This tool has been developed by Center for computational mass spectroscopy at UCSD. In addition to InsPect this center has developed number of other mass spectrometry analysis tools such as PepNovo for automated de novo sequencing of mass spectrometry data in the absence of sequence database.
Identify and quantify proteins in complex biological samples using Thermo Scientific™ Proteome Discoverer™ software. Proteome Discoverer software simplifies a wide range of proteomics workflows, from protein and peptide identification to PTM analysis to isobaric mass tagging and both SILAC and label-free quantitation. It supports multiple database search algorithms (SEQUEST, Z-Core, Mascot, and Byonic) and multiple dissociation techniques (CID, HCD, ETD, and EThcD) for more comprehensive analyses.
- Top-down and bottom-up search capabilities
- Modification Fine Control™ for fast and accurate identification, even with 20+ types of modification
- Wildcard Search™ for unknown modifications
- Intact glycopeptide identification
- Scoring models for all types of fragmentation spectra: CID, TOF-TOF, QTOF HCD, ETD/ECD, and EThcD
- Sequence variant search and modification site localization
performs LC-MS/MS data analysis and statistics according to the experimental design. Following the identification of peptides with MS/MS spectra, the resulting peptide sequences are used to determine the original protein components of the samples.
- Peptide/Protein identification
- de novo sequencing
- Database search
- Post-translational modification (PTM) search with 500+ modification
- Sequence variant and mutation search
- Protein quantification in complex biological samples
- Label-free
- Label-based: TMT (MS2, MS3) / iTRAQ, SILAC, 18O labeling, ICAT
- Supporting fragmentation types
- CID, HCD, ETD/ECD, EThcD, IRMPD, and UVPD
MaxQuant is a quantitative proteomics software package designed for analyzing large-scale mass-spectrometric data sets. It supports all main labeling techniques like SILAC, Di-methyl, TMT and iTRAQ as well as label-free quantification. Also measured spectra of various vendors - Thermo Fisher Scientific, Bruker Daltonics, AB Sciex and Agilent Technologies - can be processed using MaxQuant. The software is easy to handle and thus enables analyses of complex data sets on desktop machines by any researcher wishing to employ proteomics data.
Global Post Translational Analysis; Phospho-Proteome, Ubiquitome & BioID
Phospho-Proteome
Service Description:
1) Sample Preperation (Tissue lysis and Trypsinization):
Will require cell pellet or animal tissue. We Approximatly 1-10 mg of proteins are extracted from the samples using the Guanidine based protein extraction method.Proteins are denatured in 8M urea buffer and digested with trypsin. Peptide are purified using solid phase extraction and quantified using BCS assay for Phophopeptide enrichment.
2) Phosphopeptide Enrichment:
The Thermo Scientific™ High-Select™ Fe-NTA Phosphopeptide Enrichment Kit enables efficient enrichment of phosphorylated peptides from complex and fractionated protein digests for analysis by mass spectrometry (MS). (This product replaces Cat. No. 88300.)
Features of the Fe-NTA Phosphopeptide Enrichment Kit:
• Convenient—pre-formulated buffers for ease-of-use and spin-column format to enable parallel processing of multiple samples
• High binding capacity—each column enriches up to 150 μg of phosphopeptides
• High specificity—recovers phosphopeptides with > 90% selectivity
• Superior recovery—enriches more total and unique phosphopeptides than other commercially available resins
• High coverage—recovers peptides with single and multiple phosphorylation sites
The High-Select Fe-NTA Phosphopeptide Enrichment Kit enriches phosphopeptides from complex samples using iron-chelate resin in spin columns. Each column can enrich phosphopeptides from 0.5 to 5 mg of total protein digest as starting sample. Together with pre-formulated buffers, the improved and simplified procedure enriches up to 150 µg of phosphopeptides with greater than 90% selectivity in less than 45 minutes.
3) LC-MS Analysis:
The enriched peptides are run on a optimized 2 hour LC-MS run using a 25 cm C18 (BCH 1.7 micron) column using HCD fragmentation. Additional ETD fragmentation can be requested for additional charges. ETD fragmentation can increase the number of phosphopeptide detected in the samples.
Figures
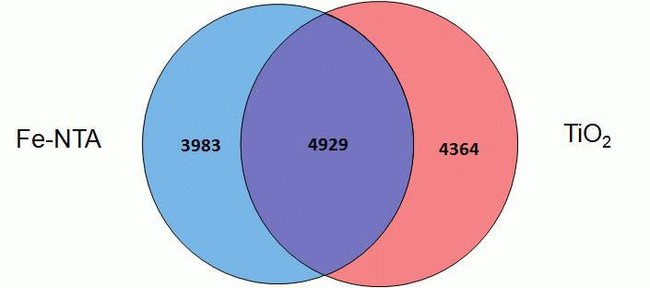
Fe-NTA and TiO2 resins enrich a complementary set of phosphopeptides
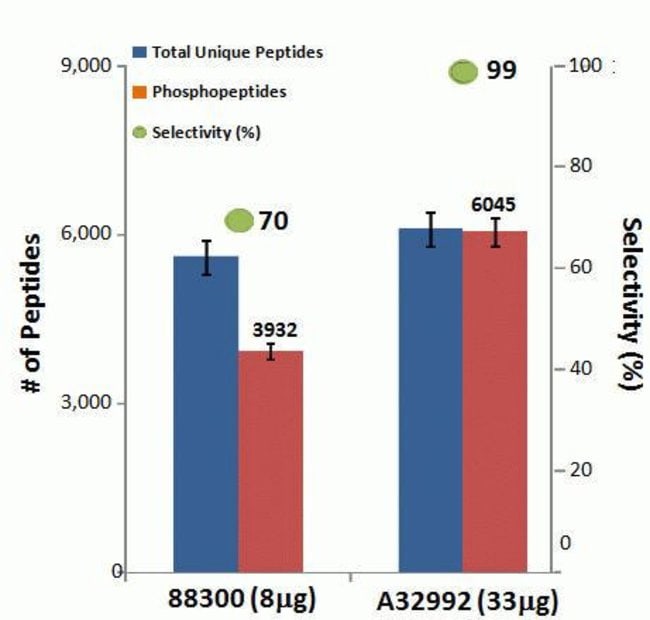
High-Select Fe-NTA Phosphopeptide Enrichment Kit improves selectivity and yield
BioID
Other Types of Analysis
Protein Quantification in Complex Protein Mixtures
For mass spectrometry base protein quantification of a given proteome, the proteome is digested using a protease and subsequently the resulting peptides are analyzed and quantified using a mass spectrometer. There are two major approaches to protein quantification:
- Relative quantification of a protein from one sample top another: In this approach the peptides can be either isotopically labeled during cell growth (SILAC, N15 & O18) or post protease digest labeled (iTRAQ, iCAT) to differentiate the samples for the quantification purpose. Also SWATH.
- Absolute quantification of a protein or proteins in a sample: In the AQUA approach proteolytic peptides of a protein are first identified and characterized. From the characterized peptides a few sequences are selected and chemically synthesized using isotopically labeled amino acids, to differentiate these peptides from their native forms, and are spiked into the proteolytic digest to be used as internal standards for the peptide quantification.
Post Translational Modifications (PTM)
Post translational modifications are important in biological processes and mass spectrometers are becoming the instruments of choice for the detection and characterization of PTMs. Essentially mass spectrometers have the capability to characterize most fixed mass modification that are stable in proteins. These include protein acetylation, ubiquitination, methylation and phosphorylation. The most important consideration in characterizing PTMs is their frequency of occurrence. Since most PTMs are rare events specific biochemical enrichment strategies need to be used to facilitate their identification and characterization. For example TiO2 and metal based resins (IMAC) are commonly used for the enrichment of phospho peptides before mass spectrometry analysis. PTM based project should be well researched and discussed to achieve the best outcome.